Xenium mouse brain
In this notebook, we will use ovrlpy to investigate the Xenium’s mouse brain dataset.
We want to create a signal embedding of the transcriptome, and a vertical signal incoherence map to identify locations with a high risk of containing spatial doublets.
We want to create a signal embedding of the transcriptome, and a vertical signal incoherence map to identify locations with a high risk of containing spatial doublets.
Settings and Imports
First we import relevant analysis packages and set the paths to the data files.
from pathlib import Path
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import ovrlpy
data_path = Path(
"/dh-projects/ag-ishaque/raw_data/tiesmeys-ovrlpy/Xenium-brain-2024/replicate1"
)
signature_matrix_file = Path(
"/dh-projects/ag-ishaque/raw_data/Xenium-benchmark/scRNAseq/trimmed_means.csv"
)
result_folder = Path("results")
result_folder.mkdir(exist_ok=True, parents=True)
Loading the data
Now, we want to load the data and prepare it for analysis.
coordinate_df = ovrlpy.io.read_Xenium(data_path / "transcripts.parquet")
coordinate_df.head()
| gene | x | y | z | |
|---|---|---|---|---|
| 0 | Bhlhe40 | 4843.045898 | 6427.729980 | 19.068869 |
| 1 | Parm1 | 4844.632812 | 6223.182617 | 18.520161 |
| 2 | Bhlhe40 | 4842.943359 | 6478.310547 | 18.500109 |
| 3 | Lyz2 | 4843.941406 | 6344.550293 | 15.016154 |
| 4 | Dkk3 | 4843.162598 | 6632.111816 | 15.394680 |
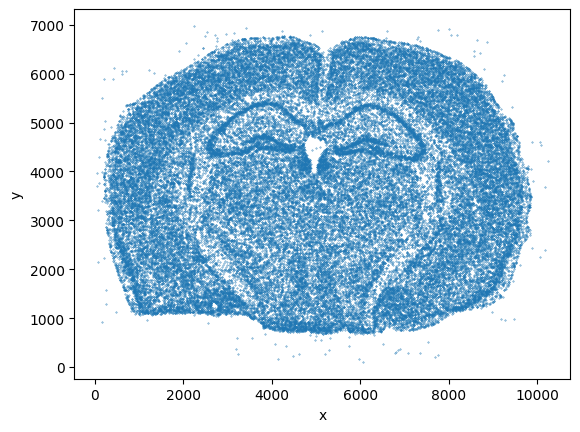
Let’s get a quick overview of the tissue
_ = coordinate_df[::1000].plot.scatter(x="x", y="y", s=0.1)
Running the ovrlpy pipeline
ovrlpy provides a convenience function run to run the entire pipeline.
The function creates a signal integrity map, a signal strength map and a Visualizer obejcet to visualize the results.
signal_integrity, signal_strength, visualizer = ovrlpy.run(
df=coordinate_df, cell_diameter=10, n_expected_celltypes=30, n_workers=8
)
Running vertical adjustment
Creating gene expression embeddings for visualization:
Analyzing in 3d mode:
determining pseudocells:
found 62341 pseudocells
sampling expression:
100%|██████████| 88/88 [02:21<00:00, 1.61s/it]
Modeling 30 pseudo-celltype clusters;
Creating signal integrity map:
94%|█████████▍| 296/315 [06:33<00:21, 1.15s/it]/dh-projects/ag-ishaque/analysis/muellni/envs/ovrlpy/lib/python3.12/site-packages/ovrlpy/_utils.py:397: RuntimeWarning: invalid value encountered in divide
spatial_patch_cosine_similarity[patch_signal_mask] = np.sum(
100%|██████████| 315/315 [06:41<00:00, 1.28s/it]
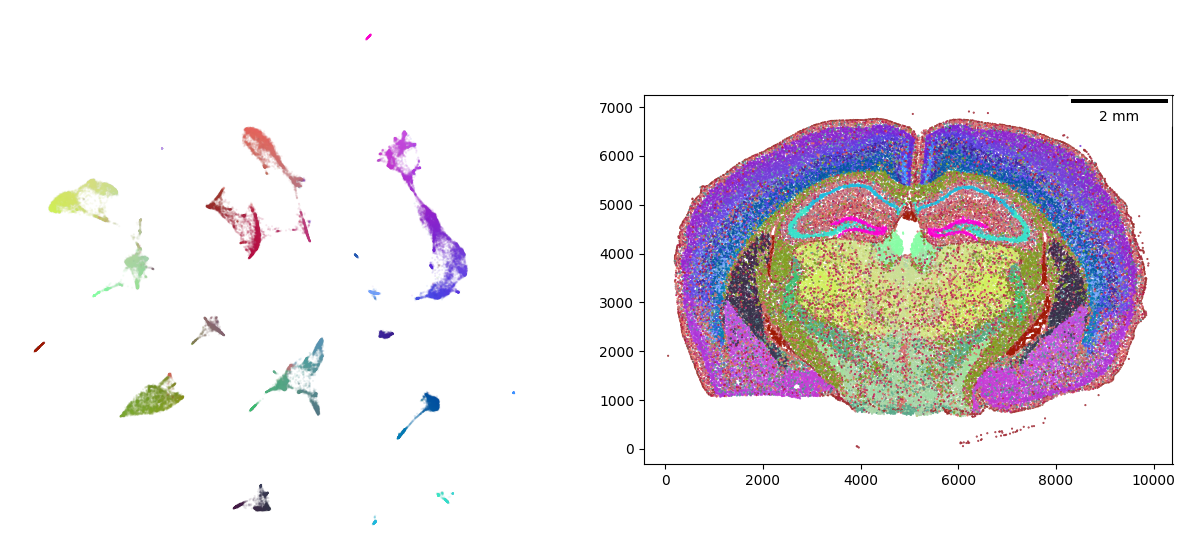
Visualizing results
The visualizer object has a plotting method to show the embeddings of the sampled gene expression signal.
visualizer.plot_fit()
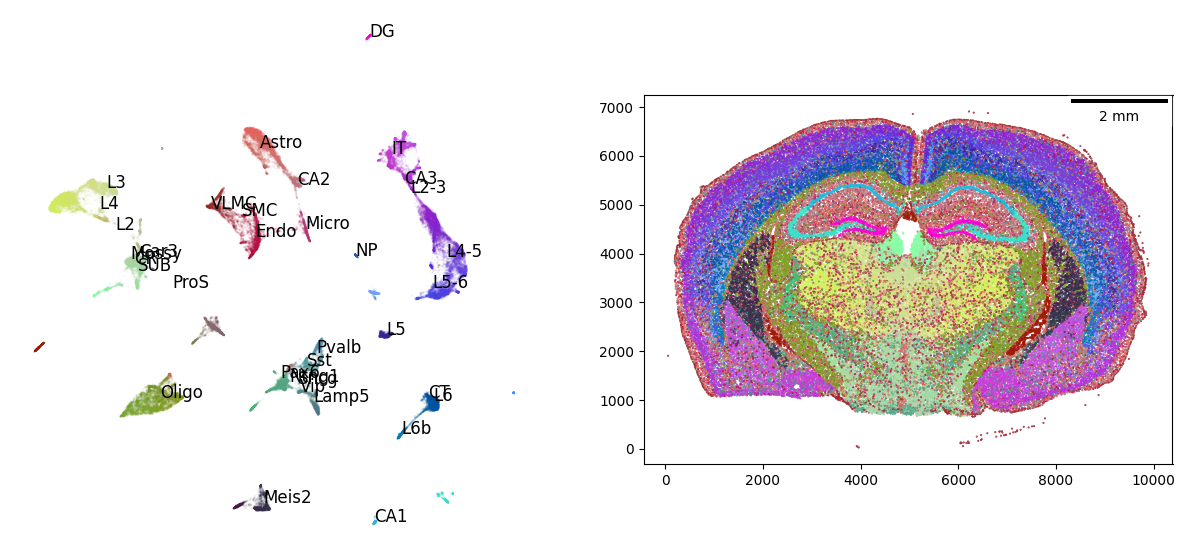
We can annotate the UMAP using external, single-cell derived cell type signatures to help interpret the cell type clusters in the gene-expression embedding:
signatures = pd.read_csv(signature_matrix_file, index_col=0).T.loc[
:, lambda df: df.columns.isin(visualizer.genes)
]
signatures = signatures.groupby(
lambda x: x.split("_")[1].split(" ")[0].split("-")[0]
).mean()
signatures.index = signatures.index.str.replace("/", "-")
signatures.index.name = "celltype"
signatures.iloc[:5, :5]
| feature | Sox17 | Col19a1 | 2010300C02Rik | Satb2 | Nrp2 |
|---|---|---|---|---|---|
| celltype | |||||
| Astro | 0.0 | 0.000000 | 0.000000 | 0.000000 | 1.316010 |
| CA1 | 0.0 | 0.000000 | 7.463820 | 2.937198 | 0.693990 |
| CA2 | 0.0 | 0.000000 | 9.586734 | 0.000000 | 2.942782 |
| CA3 | 0.0 | 0.051941 | 6.305638 | 0.000000 | 6.810690 |
| CR | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 |
visualizer.fit_signatures(signatures)
visualizer.plot_fit()
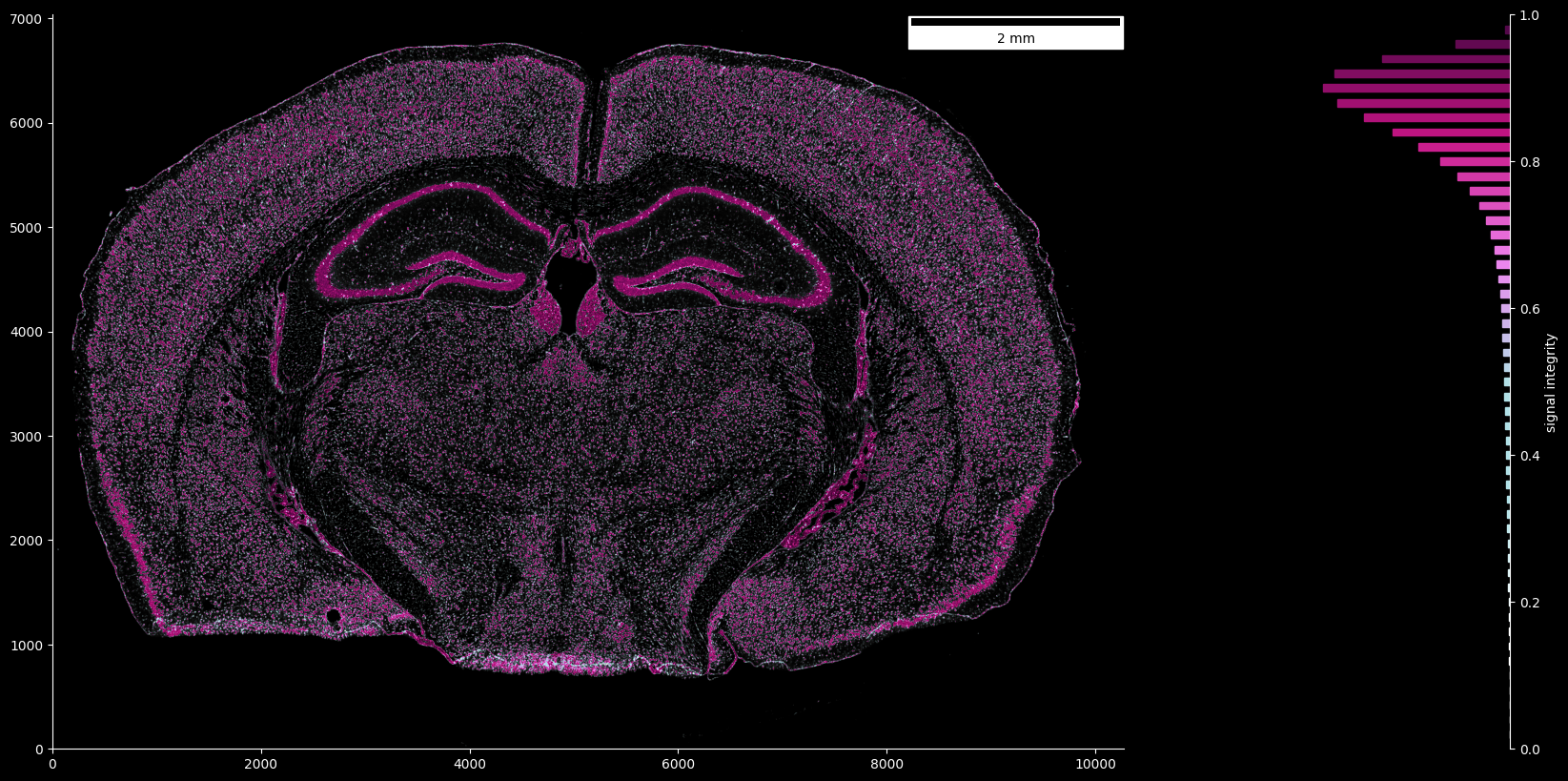
In the same way, the signal integrity map can be visualized, where visualization is cut off at regions below a certain signal strength threshold:
fig, ax = ovrlpy.plot_signal_integrity(
signal_integrity, signal_strength, signal_threshold=3
)
Detecting doublets
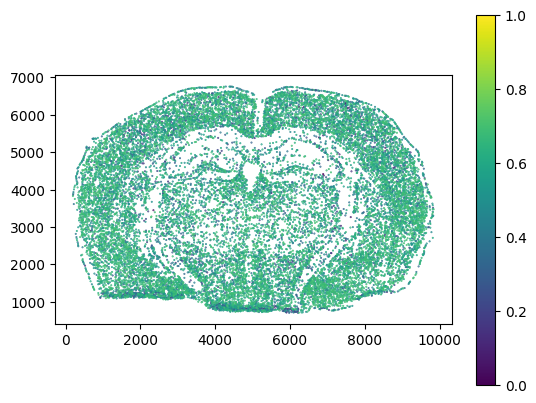
We can detect individual doublet events with ovrlpy, again setting a signal strength threshold to filter out low-transcript regions:
doublet_df = ovrlpy.detect_doublets(
signal_integrity, signal_strength, minimum_signal_strength=3, integrity_sigma=2
)
_ = plt.scatter(
doublet_df["x"],
doublet_df["y"],
c=doublet_df["integrity"],
s=0.2,
cmap="viridis",
vmin=0,
vmax=1,
)
_ = plt.gca().set_aspect("equal")
_ = plt.colorbar()
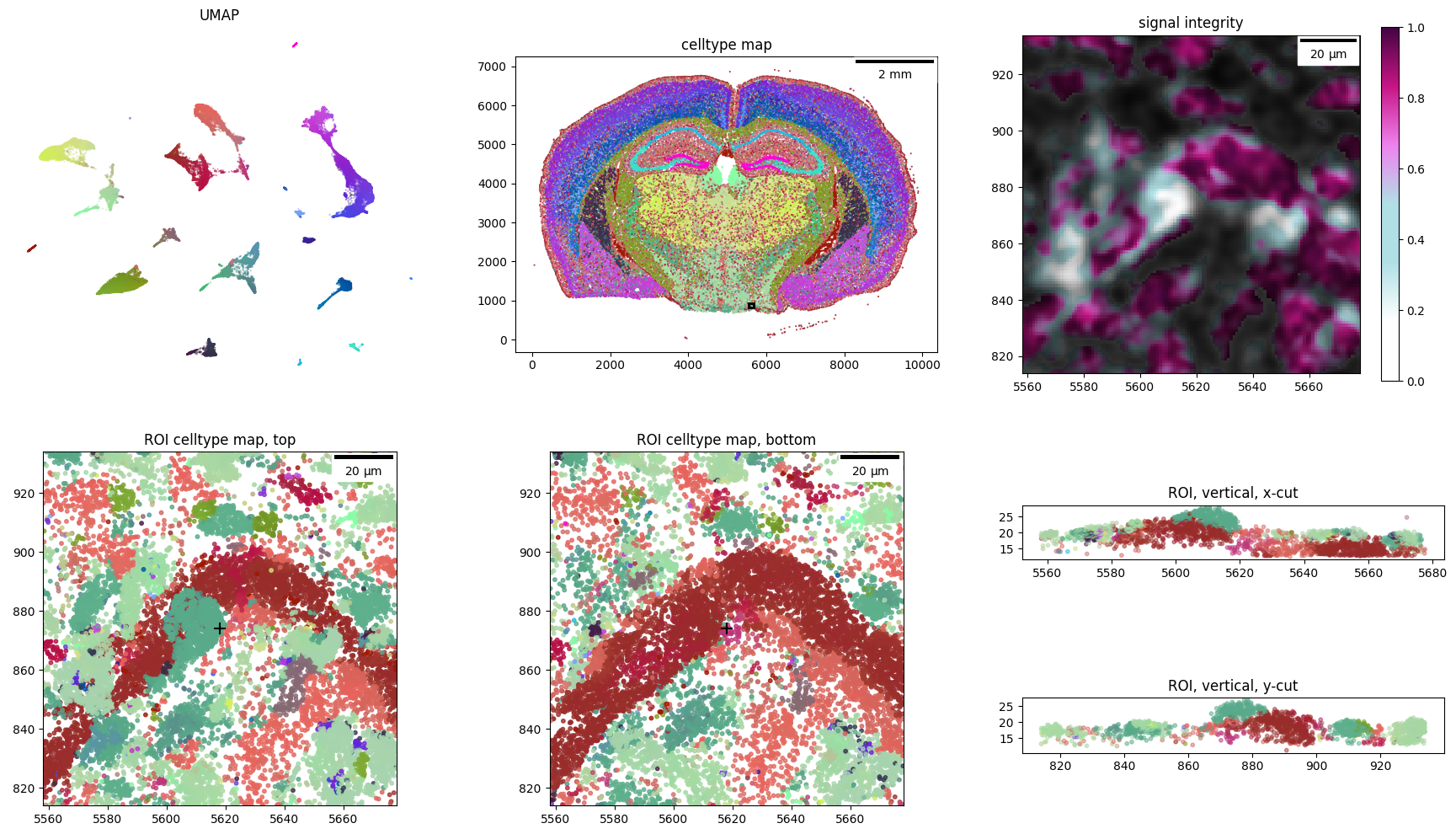
Having sampled regions of potential doublets, we can visualize them as close-up transcriptome molecule clouds through the Visualizer’s learned color embeddings - by providing their (x, y) locations to ovrlpy.plot_region_of_interest
doublet_case = 0
x, y = doublet_df.loc[doublet_case, ["x", "y"]]
_ = ovrlpy.plot_region_of_interest(
x, y, coordinate_df, visualizer, signal_integrity, signal_strength, window_size=60
)
/dh-projects/ag-ishaque/analysis/muellni/envs/ovrlpy/lib/python3.12/site-packages/sklearn/base.py:493: UserWarning: X does not have valid feature names, but PCA was fitted with feature names
warnings.warn(
Other functionality
Furthermore, we can save the visualizer object to file for later use leveraging the pickle module
import pickle
with open("my_analysis.pickle", "wb") as file:
pickle.dump(visualizer, file)
… and easily reload it if needed.
with open("my_analysis.pickle", "rb") as file:
visualizer = pickle.load(file)
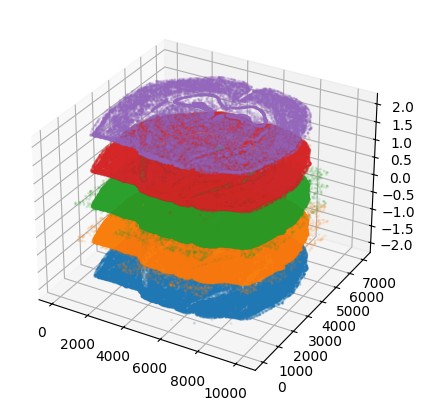
Additionally, the analysis has produced a global z-level adjustment of the transcriptome coordinates, which can be used to create a z-stack of adjacent, aligned sections in silico:
plt.figure(figsize=(20, 5))
ax = plt.subplot(111, projection="3d")
for i in range(-2, 3):
subset = coordinate_df[(coordinate_df.z - coordinate_df.z_delim).between(i, i + 1)]
ax.scatter(
subset.x[::100],
subset.y[::100],
np.zeros(1 + (len(subset) // 100)) + i,
s=1,
alpha=0.1,
)